Glutamate in Pre-Synaptic Endings |
|
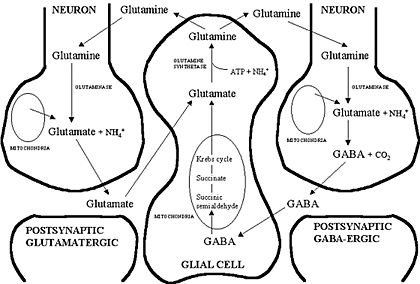
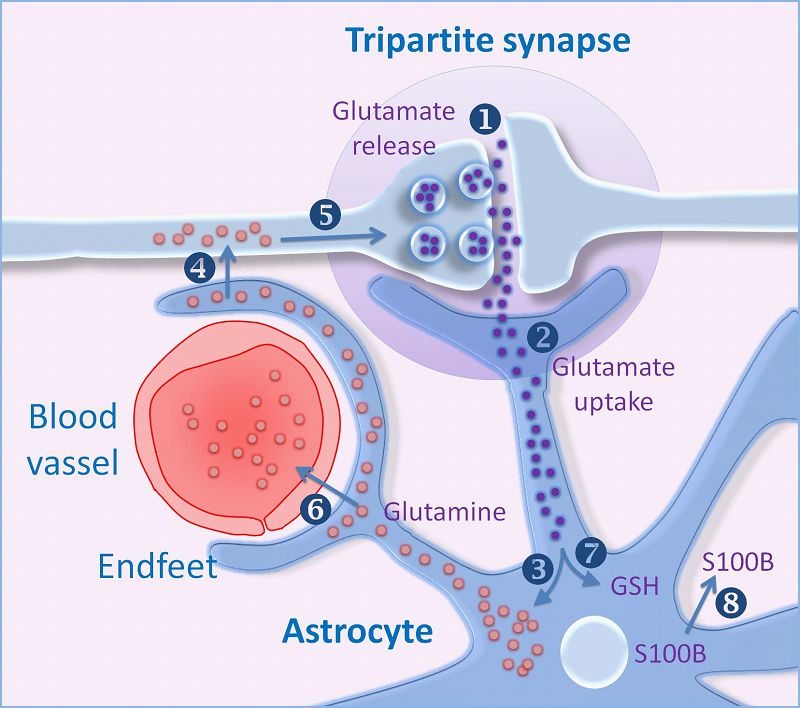
The amino acid Glutamate is synthesised from glutamine and stored within vesicles in synaptic boutons. After its release into the synapse glutamate is transported into astrocytes which convert it into glutamine, which is recycled by the nerve endings. This is the basis of the Glutamine Cycle. |
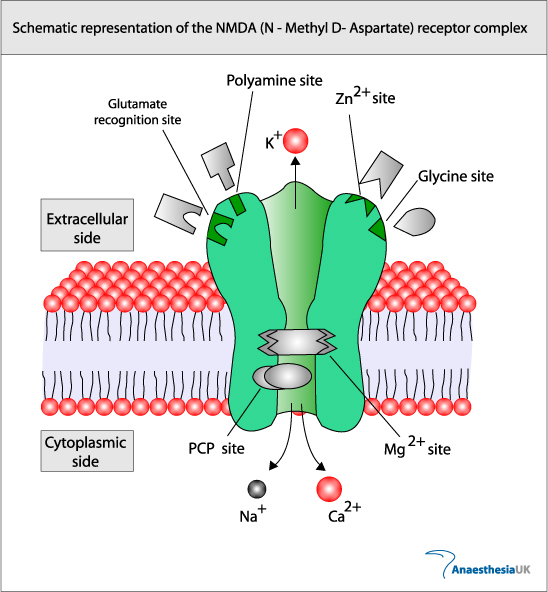
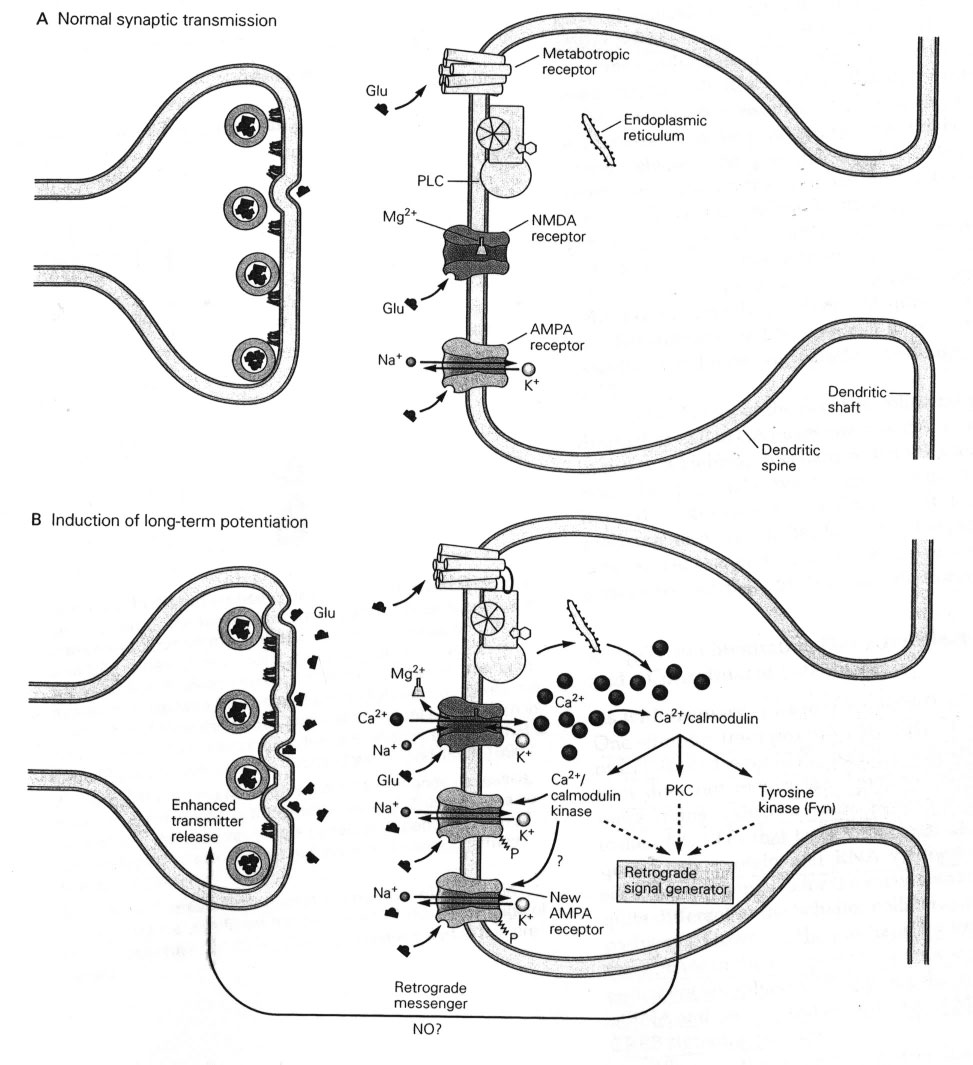
Glutamate Synthesis Neurons synthesise glutamate from glutamine. Glutamate is synthesised within glutamatergic nerve endings, but after release, it needs to be removed from the synapse quickly because accumulation of extracellular glutamate is associated with neuronal toxicity (see below). The glutamate-glutamine cycle The glutamate-glutamine cycle is recognised as an important metabolic pathway for mopping up excess glutamate from the synapse. It is a mechanism whereby glutamate released at synapses can transported into adjacent astrocytes (and neurones) using a family of transporters - the Excitatory Amino Acid Transporters (EAAT) - and converted to glutamine within the astrocyte. This glutamine can be recycled and passed back into the nerve endings for the synthesis of glutamate. Excess glutamine is transported away from the brain in the blood stream. GABA and Glutamine A similar process occurs for GABA (right). Glutamate Transport into Synaptic Vesicles After it is synthesised in the nerve endings, glutamate is transported into synaptic vesicles. Glutamate concentration in the vesicle increases to many times that of the cytoplasm. The high concentration within the vesicle is produced by an ATP-dependent proton transport system that lowers the pH of the synaptic vesicle and carries the negatively charged glutamate ions into the vesicle. The transporter involved is a family of proteins called the Vesicular Glutamate Transporters (VGLUT).
|
 * *
 * * |